SECTIONS
- Introduction
- Properties of tritium
- Complexity of tritium and its species in the nuclear industry
- Problems in treating tritium
- Tritium and tritiated water separation processes
- Conclusions
Introduction
This article deals with tritium, its different forms in radioactive effluents and an exposition of different technologies for its possible treatment. This treatment is today especially complex in the nuclear industry. Tritium is produced by fission reactions in a nuclear reactor and may be in the form of a gas, as tritiated water molecules or its ions [31H]+ and [O31H]–. A specific case was the treatment of the tritium problem in the Fukushima accident. The company treating the liquid effluent from Fukushima uses a filtering system to clean the thousands of tons of radioactive water the plant generates each day, as a result of the accident caused by the earthquake and tsunami of March 2011.
However, tritium is the only one of the radioactive isotopes that the filtering system is unable to remove. The International Atomic Energy Agency (IAEA) considers this an acceptable practice in such a context. The Japanese Ministry of Economy, Commerce and Industry requested ideas for the treatment of this tritiated water and received proposals from several companies and a university. Finally, the committee determined that controlled emptying of tritium to the sea will cost some 3.4 billion yen (€27.5 million) and take almost seven and a half years. What are the properties of tritium that have led to adopting its gradual evacuation to the sea as the most plausible solution? Why is its separation by filtration extremely complex? Why is it usually found in the primary circuit waters of a nuclear reactor? At what points in the nuclear process does the problem of tritium arise?
In September 1976, the IAEA organised a Technical Committee on the separation, storage and disposal of gaseous radionuclides from atmospheric effluents. The meeting reviewed current technology and practices for controlling gaseous emissions from fuel rework activities, and later published a report, IAEA-209. The Committee noted the need for cooperation in the area of the management of gaseous waste and recommended the technology and techniques for separating and storing all long-term isotopes be studied in depth, with special attention to iodine, the noble gases and tritium.
To comply with this recommendation, the IAEA has held meetings of experts responsible for examining the problems in question. In this article, different types of actions proposed for the particular treatment of tritium as a gas and in the form of tritiated water will be discussed.
Properties of tritium
Tritium is an isotope of hydrogen with a mass of 3.01605g/mol and is the only radioactive one of the three isotopes. Its half-life is 12.33 years; 1 g of tritium has an activity of 9619 Ci and disintegrates as below:
13H → 23He + e– + ν + 18.6 KeV
Tritium has the same chemical behaviour as deuterium and protium, as the chemical properties depend on the outer electrons which is why it is difficult to chemically separate it from other isotopes. On the other hand, it emits only beta radiation which facilitates protection systems against it. The risk from radioactive substances lies not only in their ability to irradiate externally to the person, but also especially when a person ingests a radioactive product. This means it emits within the body with the consequent irradiation to the cellular, bones and lymphatic system and, in general, to the entire internal organism. From this point of view, the introduction of tritium in the organism due to contamination is a serious problem.
At the industrial level, problems separating the different tritiated species are determined by temperatures close to the liquid-gas transition, resulting in long separation columns and extreme cryogenic temperatures (table 1).
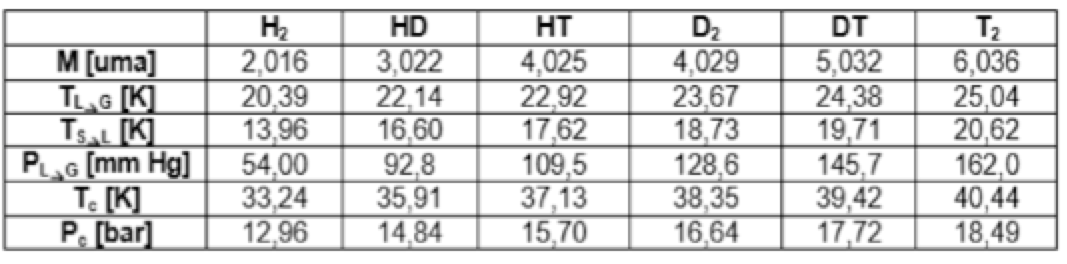
Table 1. Basic thermo-isotopic properties (CDTI)
Complexity of tritium and its species in the nuclear industry
The problem of tritium gas is part of a more complex problem that encompasses different gaseous species such as 85Kr, 14C, 3H and 129I. When reworking nuclear fuel, the current practice is to discharge a significant part of the fission products contained in the fuel – 85Kr, 14C and 3H, as well as a smaller fraction of 129I – into the environment. An annual emission estimate of these three nuclides: 85Kr, 3H and 129I was made by calculating 5.108 Ci , 7.5.107Ci and 6000 Ci. In the nuclear fuel cycle, reprocessing plants are the main cause of krypton-85 and iodine-129 emissions. Carbon-14 and tritium can be emitted in appreciable quantities both by reactors and by reprocessing plants. The method of emission, dilution and evacuation currently followed by tritium, krypton-85 and carbon-14 exposes members of the public to a small fraction of the variation in natural background radiation, well below the limits prescribed by internationally accepted radiation protection standards. Krypton-85 (with a half-life of 10.76 years) is a gaseous product produced during fission. It is initially confined in fuel rods. Due to cracks in the sheath or small corrosion cracks, up to 1% of the gas can escape and partly reach the primary circuit by dissolving in the refrigerant. During irradiation, over 99% of it remains in these fuel elements until they are broken and dissolved during reworking. Finally, the gas is released and obligatorily treated in gas treatment systems. Then it passes entirely into the waste gas system. About 330,000 Ci of krypton-85 are emitted per gigawatt electrical year (GWe.year) generated for light water reactor (LWR) fuels, and 580,000 Ci/GWe.year for high temperature gas-cooled reactor fuels (HTGR).
The difference between emissions is due to the different amounts of krypton-85 produced by the fission of uranium-235 and uranium-233 used respectively in these two types of reactors. PWRs, such as those at Ascó and Vandellós, use a small proportion of between 3-4%235U. For liquid metal cooled fast breeder reactors (LMFBR), radioactive emission is estimated at 1.2-2.1 x 105Ci/GWe.year, approximately. To date, virtually all of the krypton-85 from reworking the fuel has been emitted into the atmosphere.
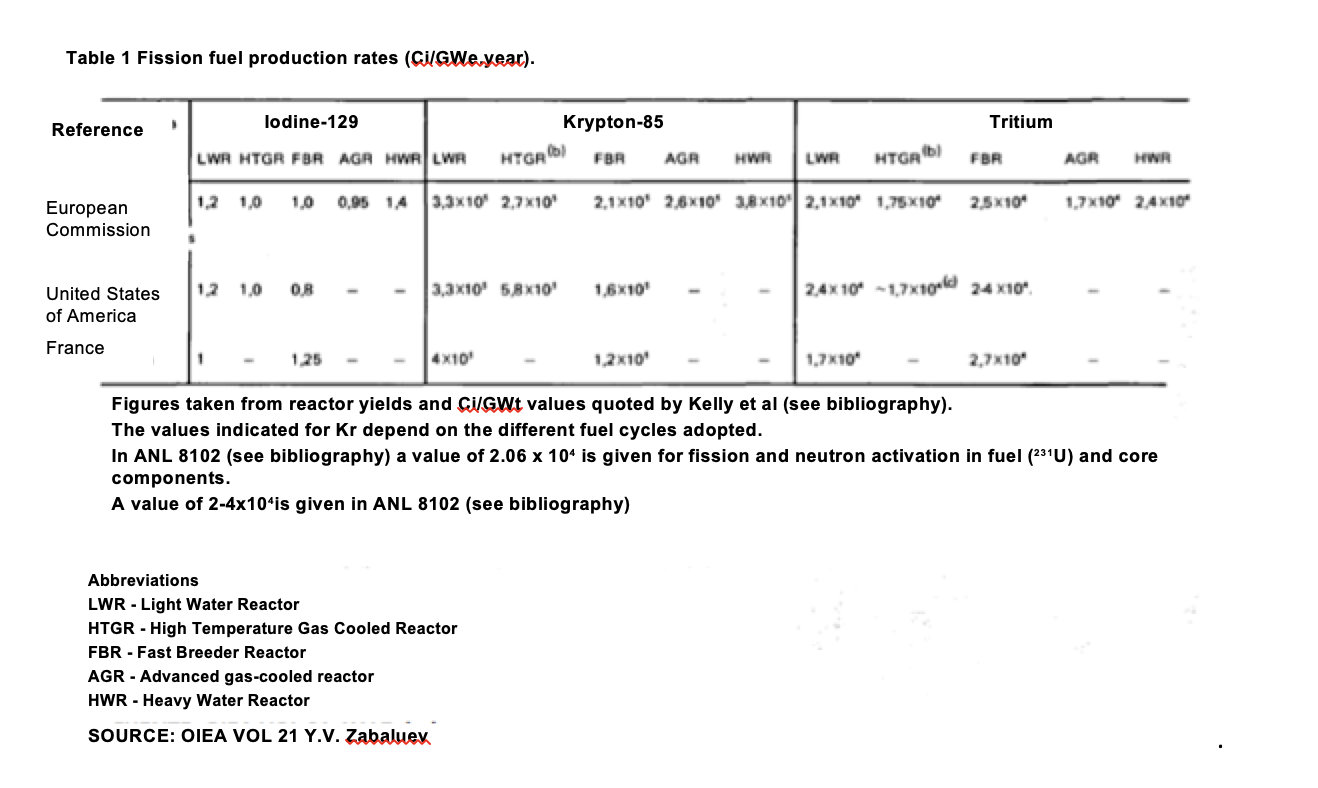
Table 2. OIEA vol 21. Y.V.Zabaluev
All the iodine-129 (half-life 1.7 X 107 years) is a direct fission reaction product formed inside a zirconium alloy sheath. It is retained almost entirely in the fuel until its dissolution. The formation rate is approximately 1.0 Ci/GWe.year for all types of reactors (Table 1). In the fuel reworking processes where the components are mechanically cut, over 98% of the iodine goes into the waste gas system after the fuel dissolves, and measurements are usually taken to separate it from these gases to limit the iodine-131 emissions. For example, it is retained with activated carbon and subsequently fixated with silver nitrate in specific filters.
Tritium (half-life of 12.3 years and emitting only βradiation) is formed in nuclear fuels primarily by ternary fission, at a rate of 200,000-400,000 Ci/GWe.year (Table 1). It is also formed by neutron activation of a series of light elements present as impurities or as components of the fuel, coolant, moderator, sheaths and other nuclear materials. It is currently possible to obtain it from existing fission reactors that use heavy water (D2O) as a moderator (e.g. Candu), as they produce T(tritium) when deuterium (D) captures a neutron. Heavy water from these reactors must be “cleaned” regularly, so that they represent a more or less regular source of tritium, e.g. in Canada. In PWR reactors, tritium is produced by interaction of Lithium-6 with neutrons, according to the reaction:
![]()
Lithium-6 is used in the form of LiOH with the ability to regulate the pH in solution. Also, boron-10, used as a neutron absorber, in the form of H3BO3, thus regulating the reactivity of the nucleus. Natural boron contains 20% boron-10 and approximately 80% boron-11. Boron-10 has a high cross-section for absorbing low-energy (thermal) neutrons. The addition of boric acid, in addition to the refrigerant circulating through the reactor, reduces the probability of a neutron fissuring a uranium atom. Changes in boric acid concentration effectively regulate the fission rate taking place in the reactor. This method is used only in pressurised water reactors (PWR). Boron also dissolves in spent fuel pools containing spent uranium bars. The concentration is high enough to keep the neutron reactivity rate to a minimum. Boric acid was poured onto reactor 4 of the Chernobyl reactor after the accident, to prevent another reaction from occurring. In general, boron-10 absorbs the neutron forming a boron-11 atom in an excited state which turns into lithium (Li-7); this too is unstable and decomposes into tritium and helium. Lithium-6, by absorption of neutrons, also forms Li-7 before turning into tritium. In general, the most commonly used neutron poisons are part of the sources of tritium.
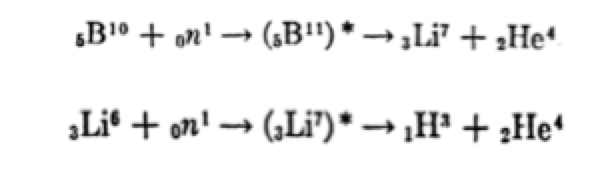
Tritium appears in ternary fissions as a result of the following reactions:
235U + n → X1 + X2 + H3
239Pu + n → X1 + X2 + H3
Both the moderator and refrigerant in PWRs operate at high temperatures and pressures, and there is also the possibility of tritium exchange between the two by diffusion during normal operation of the plant and by mixing when stopped. The International Commission on Radiological Protection (ICRP) limits the dose for workers to a 5-year average of 20 mSv per year. The high contribution of tritium to the total dose has encouraged the study, development and optimisation of technologies for tritium control for reactors in operation as well as for advanced designs. The tritium values for PWRs are around 330 Bq/g and for I-131 are around 9 Bq/g.
The tritium generated in nuclear reactions is in the form of tritium gas [31H]2 or mostly forming part of the water molecule in the form:
31H-OH (T-OH) ;H-O31H (H-OT); [31H]2O(T2O)
The reaction of tritium with oxygen forms tritiated water: T2O
2T2 + O2 → 2 T2O
Air + T2 → T2O + T2O2 + NO + NO2
T2 + CO2 → T2O + CO
Chemical species present in the aqueous medium:
H2O ; T2O ; HTO; T2O+; T3O+; OT–; T2O2
A fundamental property of tritium is the ease of exchange with protium. The equilibrium state depends on the acid-base reactions with the other chemical species in solution, with the pH of the solution being decisive.
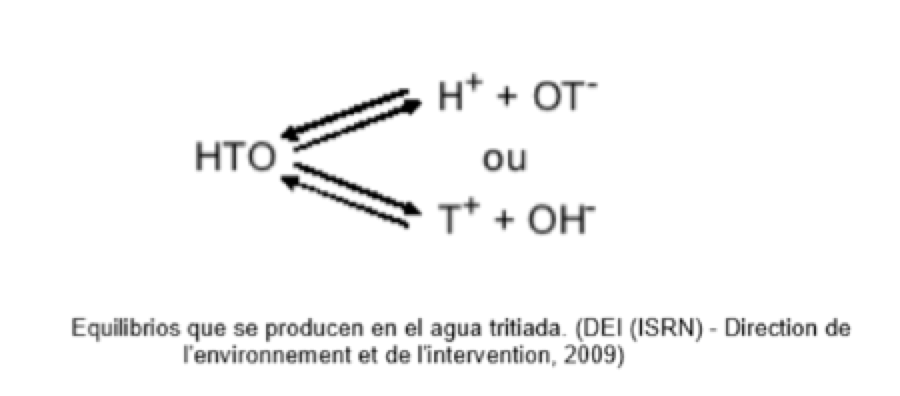
This ease for tritium to form protium results in a particularly difficult problem in the regeneration of boric acid.
In aqueous solution, the H3BO3 forms the tetrahydroxyborate complex:
H3(BO3) + 2H2O → [B(OH)4]− + H3O+ (1)
According to this reaction, boric acid would then behave not as a Brønsted acid (a proton donor) but as a Lewis acid that interacts with the water molecule as a hydroxyl anion acceptor. Some authors (Perelygin and Chistyakov, 2006) postulate the behaviour of boric acid as a tribasic Brønsted acid that reacts with water in successive stages:
H3(BO3) + H2O → [BO(OH)2]− + H3O+ (2)
[BO(OH)2]− + H2O → [BO2(OH)]2− + H3O+
[BO2(OH)]2− + H2O → (BO3)3− + H3O+
while others describe the dissociation in the usual form of acid anion and hydronium cation, (Nakai et al., 1988):
H3(BO3) + H2O → [H2(BO3)]− + H3O+
Due to the above, discrepancies are observed in the interpretation of the origin of the acid behaviour of H3(BO3) solutions. Studies of strongly alkaline solutions by Raman spectroscopy have shown the existence of the complex [B(OH)4]−, (Jolly, 1984), which supports the hypothesis of reaction (1), which implies that the acid behaviour is due exclusively to the separation of the hydroxyl anion from water, (Housecroft and Sharpe, 2005). For this reaction (1) the acid constant is low, Ka = 7.3 × 10-10. For reaction (2) the acid constant has the value Ka = 5.98×10−10 in H2O and Ka = 1.83 × 10−10 in D2O, (Gold and Lowe, 1968).
In any case, the reception of tritium and tritiated hydroxyl is feasible in the equilibrium of boric acid; thus, there are boric acid molecules with tritium, causing an added difficulty when trying to separate the boric acid by evaporation as well as problems in the boric acid recovery treatment by exchange resins. In the evaporation, boric acid with tritium incorporated in its molecule can be obtained, with the management problems generated by this situation.
Taking into account that the atom of protium can capture a neutron by the action of neutron flux and transform into deuterium. Deuterium and tritium can react forming other different aqueous structures. Considering that the tritium isotope is a net β emitter with a half-life of 12.3 years, it can occur in any of the molecular structures, including the gaseous species, and this makes any attempt at industrial level separation extremely difficult. In September 1976, the IAEA organised a Technical Committee on the separation, storage and disposal of gaseous radionuclides; in particular, tritium treatment systems were proposed. The different technical studies proposed worldwide for effective treatment of tritium emissions to reduce the occupational dose and possible environmental contamination from it, led to the following general objectives, among others:
1. Minimise losses of tritiated water, as well as its recovery in liquid and vapour state.
2. Replacement of highly tritiated heavy water with low tritium content heavy water.
Problems in treating tritium
Currently, the tritium generated in nuclear power plant radioactive effluents, before treatment with resins to remove low and medium activity species, is stored and released under control by regulations. In Spain, this regulation establishes a maximum tritium activity of 100 Bq/L. It must be remembered that the tritium values in the coolant can reach the order of 200,000 Bq/L. Each hydrographic basin limits the annual discharge of both coolant and industrial water. This fact leads to the provision of tritiated water storage in the plant itself in concrete tanks and dosing of the emission.
The evolution of tritium activity in gaseous effluents is parallel to that observed in the reactor coolant where the generation of this isotope in BWR reactors is due to neutron absorption (neutron poison from control rods), ternary fission and deuterium activation. Ternary fission and deuterium activation are practically constant, so the increase in tritium production is basically due to the generation of tritium within the control rods and its diffusion.
The chemical and physical properties of tritium, as well as the different combination options in the formation of water molecules, currently make large-scale industrial separation systems difficult. However, this does not mean that tritium treatment systems are not being developed. On the contrary. The number of articles and experimental processes for the treatment, capture and storage of tritiated species is increasing. This is also being expedited by the construction of fusion reactor prototypes.
Tritium and tritiated water separation processes
This section reviews different processes proposed for the separation of tritium, while considering that articles and laboratory tests on this subject are growing continuously, due to the management of tritium in fusion reactors and tritiated water management in conventional processes and nuclear power plant decommissioning.
a. Hydride formation
Hydrogen gas and therefore T2 react at high temperature with transition metals forming hydrides: scandium, yttrium, lanthanum, the actinides and especially the titanium and vanadium group elements.
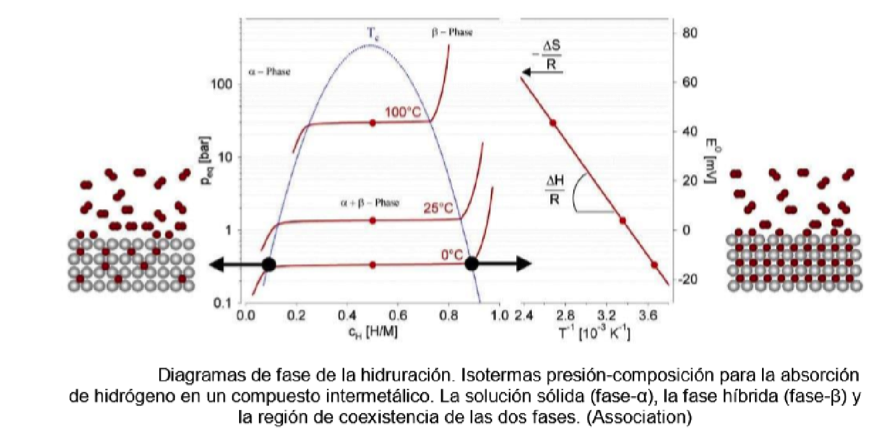
Table 3. Pressure-composition isotherms for hydride formation
The most efficient way to capture hydrogen to form hydrides is with uranium, but for reasons related to the use of uranium, the ZrCo alloy is used reversibly as follows:
Adsorption: 2ZrCo + 3T2 → 2ZrCoT3 + Q
Desorption: 2ZrCoT3 + Q → 2 ZrCo + 3T2
The problems with this compound are its instability and thermal decomposition, as follows:
2ZrCoT3 + Q → ZrCo2 + ZrX2 + + 2X2
The use of hydrides offers a possibility of containing the T2. There are operational processes developed by Professor T. Motyka for the confinement of tritium (T2) with hydrides.
b. The company, Molecular Separations, Inc (MSI), has developed a patent for a bed of particles that selectively charges tritiated water as hydration water at close to ambient temperatures. The tests were performed with a standard mixture of 126 μCi tritium/litre of water. Reductions to 25 μCi tritium/litre of water were shown using 2 x 2 m columns in series. Using wastewater samples from Hanford, a reduction from 0.3 μCi to 0.07μCi tritium/litre of water was achieved. The tritium absorbed by the beds can be released with a moderate increase in temperature and the beds can be re-used. A mobile bed process has been proposed to treat representative amounts of wastewater. It has also been shown that the separation system reduces tritium concentrations in cooling water to levels that allow its re-use.
c. Studies related to tritium adsorption and treatment of tritiated liquid effluents have been carried out for a few years at the Belgian Centre for Nuclear Research, SCK/CEN. Initially, the studies focused on the removal of tritium from gaseous effluents originating in reprocessing procedures. If tritium can be released from spent fuel before any aqueous operation, the most practical collection method is adsorption on molecular sieves, after an isotopic solution with hydrogen and subsequent complete transformation to tritiated water. SCK/CEN constructed an adsorption unit by oxidation of 15 m3/h with a closed regeneration system and a decontamination factor of 1000 in the total concentrations of tritiated hydrogen, and a water inlet up to 1000 parts per million per unit volume. SCK/CEN is developing an isotope separation process called ELEX based on combining water electrolysis and tritium exchange between hydrogen and water, with a hydrophobic catalyst promoting the exchange. For electrolysis under normal conditions, an elementary tritium separation factor of 11.6 was obtained with a standard deviation of 6%. Regarding exchange capacity, a hydrophobic catalyst was developed that yields an overall exchange rate constant of 9 mol/s.m3 in a countercurrent drip bed reactor for flows used at atmospheric pressure and 20°C. This pilot plant consists of two essential parts: an 80 kW water electrolyser and a 10 cm diameter drip bed column. The tritiated water feed rate is 5 L/h, which contains a tritiated aqueous phase of 3.7GBq/L of tritium activity.
d. Using the multiple bipolar electrolytic separation of hydrogen isotopes by Pd/Ag electrodes (25% Pd), the possibility of separating tritium and tritiated species from different types of effluents was demonstrated. Bipolar processes were achieved experimentally by individual cascade cells with a bipolar electrode of the same area as the others arranged in series. The factors measured for multibipolar H-D separation were close to the values measured in single-stage cell measurements; for the H-T separation, the leakage between stages reduced the measured separation factor. However, in both cases a separation of sufficient magnitude was achieved to show the viability of a real application in the tritium extraction of large volume systems with a high current density.
e. Cryogenic distillation. Required in this option is a pre-distillation step, an electrolytic process that transforms the tritiated water into H2, T2 gas molecules and deuterium, if applicable. These gases can be stored in titanium beds. This distillation process is carried out at 24K and is one of the methods tested for enrichment and separation of hydrogen isotopes on an industrial scale with a good separation factor. The disadvantages of this type of plant are the high energy cost to maintain the extreme cryogenic temperatures as well as the high content of tritium inventory.
Conclusions
Tritium causes problems of varying degrees in the treatment of effluents from nuclear reactors, regardless of the type of reactor. After treatment with resins of the different low and medium activity species in the coolant and the separation of boric acid, the reactors store remaining effluents with tritium activity. Emission to the environment depends on the criteria of the regulator; in Spain this is about 100 Bq/L, so tritiated water storage systems are installed in the plants themselves. The total volume of cooling water discharged annually by power plants is established by the river basins, depending on the type of basin and its hydrological features. Limit values of the order of 50m3 a week may be common. This results in a problem of tritiated water storage in the plant itself. The problem of tritium treatment was highlighted In the accident at Fukushima. After various technical studies, the Japanese Ministry of Economy, Commerce and Industry opted for controlled periodic release of a certain amount of tritium to the sea, with the consensus of the fishing and craft industries in the area. Tritium treatment is an object of industrial study and application, not only in the conventional nuclear (fission) industry but in new nuclear fusion reactor (ITER) projects. Currently, cryogenic distillation, preceded by electrolysis that converts the tritiated water into T2 and other hydrogen isotopes, together with storage of the gases, producing hydrides with matrices of the titanium, ZrCo type and the more than likely use of new specific cascade separation membranes to separate different tritiated species are industrially scalable technologies to address the problem. In the future, this type of technology will be necessary to deal with decommissioning tasks and managing tritium in fusion reactors.
1.- M.Rodman, D Howard “Simulation Calculations for a Catalytic Exchange/Cryogenic Distillation Hydrogen Isotope Separation Process” E.I. du Pont de Nemours & Co. Savannah River Laboratory . DP-MS-84-100.
2.-R.Sherman. “Cryogenic Hydrogen Distillation for The Fusion Fuel Cycle” Fusion Technology Vol.8 Sep 1985 pp.2175-2183.
3.-D.Murdoch. “Sulzer builds tritium removal plant”. Modern Power Engineering. April 1982. p30.
4.-T.Motyka “Hybrides for processing and Storing Tritium” Hydrogen Technology Section of the Savannah River Technology. WSRC. 2000. pp.187-195.
5.-W.J.Holstlander, T.E.Harrison, V.Goyette, J.M.Miller . “Recovery and Packaging of Tritium from Canadian Heavy Water Reactors”. Fusion Technology Vol.8. Sep 85. pp. 2473-2477.
6.-J.M.Miller, W.T.Shmayda, S.K.Sood, D.A.Spagnolo . “Technology Developments for Improved Tritium Management” . AECL-10964. June 1994.
7.-S.K.Sood, R.A.P.Sissingh, O.K.Kveton. “Removal and Inmobilization of Tritium from Ontario Hydro’s Nuclear Generating Stations”. Fusion Technology Vol.8. Sep.85.pp. 2478-2485.
8.- Y.V. Zabaluev .Gestión de los radionucleidos procedentes de efluentes gaseosos, de plantas de reelaboración.OIEA Vol. 21 nº 1
